Histórico da Farmacovigilância
De acordo com Strom, a história da regulação de medicamentos corre paralela à história dos maiores desastres envolvendo reações adversas a medicamentos. Acompanhe os marcos internacionais:
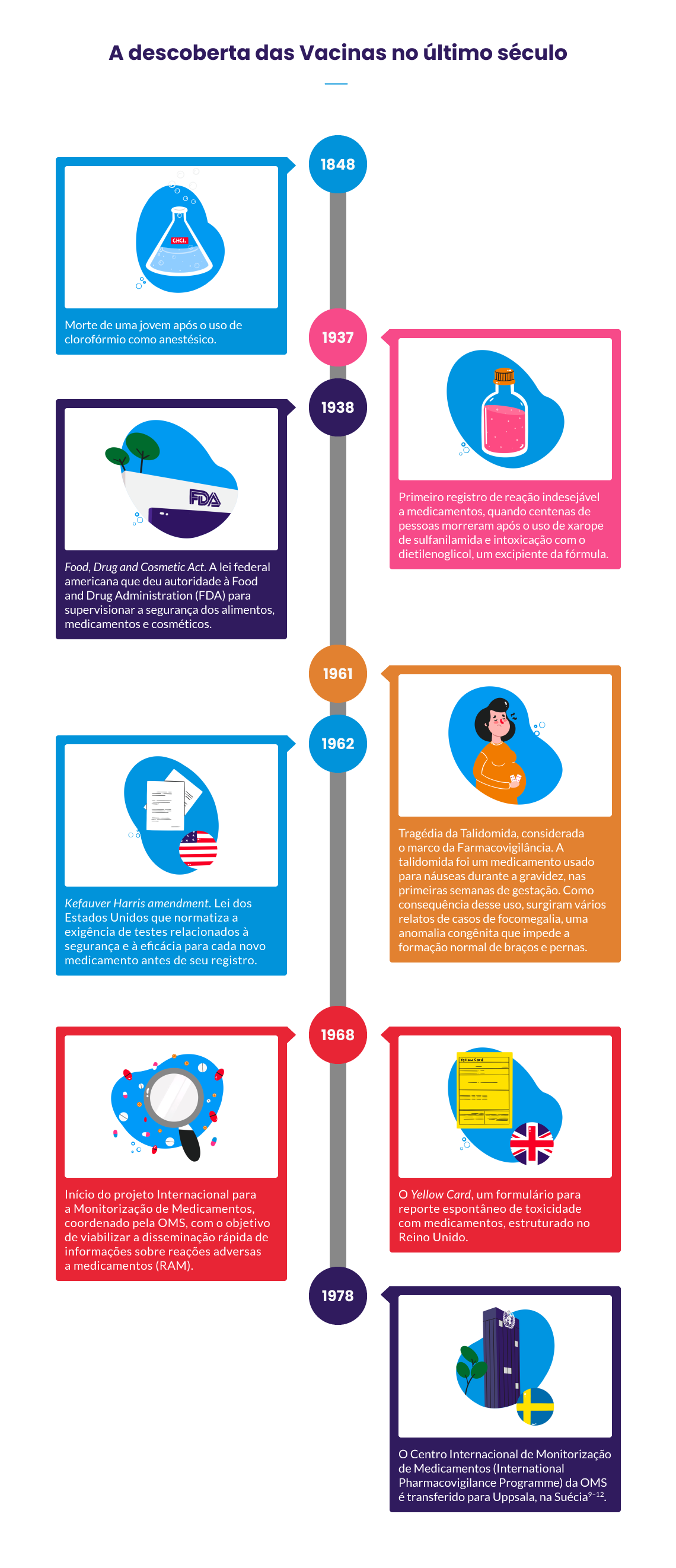
Fontes: Castro (2000); Fornasier e colab. (2018); Gomes e Reis (2006); OPAS (2011); Strom (2005); Storpirtis e colab. (2013).
No Brasil, o monitoramento da segurança dos medicamentos começa a ser regulamentado com a Lei 6.360 de 23 de setembro de 1976, que dispõe sobre a notificação de acidentes ou reações nocivas causadas por medicamentos à autoridade sanitária. Posteriormente, a Política Nacional de Medicamentos (Portaria 3.916 de 30 de outubro de 1998 - Ministério da Saúde) traz ações prioritárias de Farmacovigilância quanto ao uso racional de medicamentos. No entanto, as atividades regulatórias de farmacovigilância foram efetivamente organizadas no Brasil a partir da criação da Agência Nacional de Vigilância Sanitária (Anvisa) em 1999.
Em 2001, foi criado o Centro Nacional de Monitorização de Medicamentos (CNMM), sediado na Gerência de Farmacovigilância (GFARM) da Anvisa, após a tragédia com a meglumina, um medicamento utilizado no tratamento da leishmaniose, que provocou centenas de reações adversas fatais, reforçando a necessidade de estruturar atividades relacionadas à segurança de medicamentos no país.
A criação do CNMM propiciou a entrada do Brasil no Programa Internacional de Monitorização de Medicamentos da OMS. O Brasil é o 62º país-membro do programa.
A Rede de Centros de Informação sobre Medicamentos, vinculada à Organização Panamericana da Saúde (OPAS), reúne as informações dos Centros da América Latina e Caribe, que compõem a rede

A regulamentação dos sistemas de farmacovigilância para os produtores de medicamentos e vacinas ocorreu em 2009, a partir da Resolução nº 4, de 10 de fevereiro de 2009 - Ministério da Saúde, que estabeleceu a regulamentação dos sistemas de farmacovigilância para os produtores de vacinas - desde a notificação compulsória dos EAPV relacionados ao uso de seus produtos até o desenvolvimento de uma estrutura capaz de monitorar a segurança dos medicamentos comercializados.
Essa Resolução representou um grande avanço na vigilância pós-comercialização de medicamentos no Brasil, estabelecendo ações de farmacovigilância às indústrias, que vão desde a notificação compulsória dos EAPV relacionados ao uso de seus produtos até o desenvolvimento de uma estrutura capaz de monitorar a segurança dos medicamentos comercializados.
Em 2016, a Anvisa tornou-se membro do Conselho Internacional para Harmonização de Requisitos Técnicos para Medicamentos de Uso Humano (ICH), conselho que reúne autoridades reguladoras e associações de indústrias farmacêuticas para discutir aspectos técnicos e científicos relacionados ao registro de medicamentos. Por fim, em 2020, entrou em vigor a Resolução de Diretoria Colegiada - RDC 406 de 22 de julho de 2020, que dispõe sobre as Boas Práticas de Farmacovigilância para Detentores de Registro de Medicamento de uso humano.